cayenne: a Python package for stochastic simulations
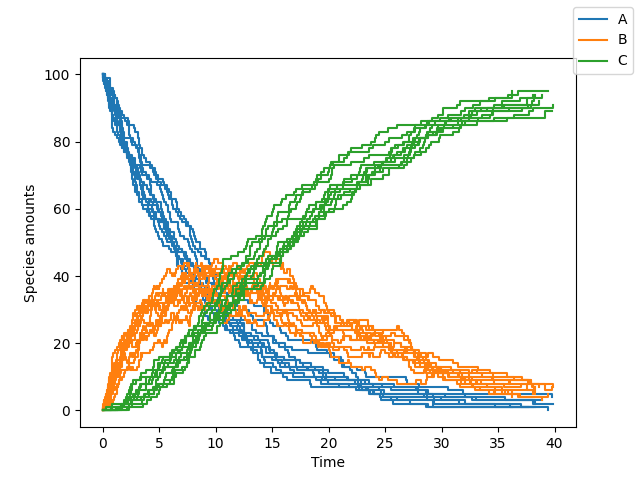
cayenne is a Python package for stochastic simulations. It offers a simple API to define models, perform stochastic simulations with them and visualize the results in a convenient manner.  ## Install Install with `pip`: ```bash $ pip install cayenne ``` ## Documentation - General: https://cayenne.readthedocs.io - Benchmark repository, comparing `cayenne` with other stochastic simulation packages: https://github.com/Heuro-labs/cayenne-benchmarks ## Usage A short summary follows, but a more detailed tutorial can be found [here](https://cayenne.readthedocs.io/en/latest/tutorial.html). You can define a model as a Python string (or a text file, see [docs](https://cayenne.readthedocs.io)). The format of this string is loosely based on the excellent [antimony](https://tellurium.readthedocs.io/en/latest/antimony.html#introduction-basics) library, which is used behind the scenes by `cayenne`. ```python from cayenne.simulation import Simulation model_str = """ const compartment comp1; comp1 = 1.0; # volume of compartment r1: A => B; k1; r2: B => C; k2; k1 = 0.11; k2 = 0.1; chem_flag = false; A = 100; B = 0; C = 0; """ sim = Simulation.load_model(model_str, "ModelString") # Run the simulation sim.simulate(max_t=40, max_iter=1000, n_rep=10) sim.plot() ```  ### Change simulation algorithm You can change the algorithm used to perform the simulation by changing the `algorithm` parameter (one of `"direct"`, `"tau_leaping"` or `"tau_adaptive"`) ```python sim.simulate(max_t=150, max_iter=1000, n_rep=10, algorithm="tau_leaping") ``` Our [benchmarks](https://github.com/Heuro-labs/cayenne-benchmarks) are summarized [below](#benchmarks), and show `direct` to be a good starting point. `tau_leaping` offers greater speed but needs specification and tuning of the `tau` hyperparameter. The `tau_adaptive` is less accurate and a work in progress. ### Run simulations in parallel You can run the simulations on multiple cores by specifying the `n_procs` parameter ```python sim.simulate(max_t=150, max_iter=1000, n_rep=10, n_procs=4) ``` ### Accessing simulation results You can access all the results or the results for a specific list of species ```python # Get all the results results = sim.results # Get results only for one or more species results.get_species(["A", "C"]) ``` You can also access the final states of all the simulation runs by ```python # Get results at the simulation endpoints final_times, final_states = results.final ``` Additionally, you can access the state a particular time point of interest $t$. `cayenne` will interpolate the value from nearby time points to give an accurate estimate. ```python # Get results at timepoint "t" t = 10.0 states = results.get_state(t) # returns a list of numpy arrays ``` <h2 id="benchmarks"> Benchmarks </h2> | | direct| tau_leaping | tau_adaptive | --- | --- |--- | --- | cayenne | :heavy_check_mark: Most accurate yet | :heavy_check_mark: Very fast but may need manual tuning| Less accurate than GillespieSSA's version| Tellurium | :exclamation: Inaccurate for 2nd order | N/A | N/A | GillespieSSA | Very slow |:exclamation: Inaccurate for initial zero counts | :exclamation: Inaccurate for initial zero counts BioSimulator.jl | :exclamation: Inaccurate interpolation | :exclamation: Inaccurate for initial zero counts | :exclamation: Inaccurate for initial zero counts ## License Copyright (c) 2018-2020, Dileep Kishore, Srikiran Chandrasekaran. Released under: Apache Software License 2.0 ## Credits - [Cython](https://cython.org/) - [antimony](https://tellurium.readthedocs.io/en/latest/antimony.html) - [pytest](https://docs.pytest.org) - [Cookiecutter](https://github.com/audreyr/cookiecutter) - [audreyr/cookiecutter-pypackage](https://github.com/audreyr/cookiecutter-pypackage) - [black](https://github.com/ambv/black) - Logo made with [logomakr](https://logomakr.com/)
